The 28 members of the Council of the EU unanimously voted to adopt the Medical Device Regulations (MDR) and In Vitro Diagnostics Regulations (IVDR), just two weeks after releasing the final texts of the regulations.
The new rules introduce deep changes in the regulatory framework for the marketing of devices and IVDs in Europe, the world’s second-largest device market.
Medical Device Regulation (MDR) final text
http://data.consilium.europa.eu/doc/document/ST-10728-2016-INIT/en/pdf
In Vitro Diagnostic Regulation (IVDR) final text
http://data.consilium.europa.eu/doc/document/ST-10729-2016-INIT/en/pdf
Adoption timelines
• EU Council has unanimously voted adoption of the MDR and IVDR on March 7, 2017.
• The European Parliament will then vote to enact the legislation in April, 2017.
• The texts will be published in the Official Journal of the European Union.
• MDR regulations will take full effect in 2020 (after a three-year transition period).
• IVDR regulations will be fully implemented in 2022 (after a 5-year transition period).
• It remains to be seen whether the UK will adopt the MDR and IVDR.
Content of the new legislation
The texts of new regulations include, among several measures:
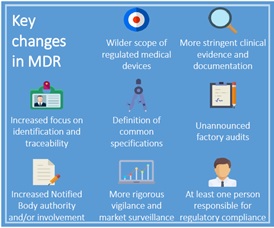
• new rules to tighten regulation by national authorities of notified bodies,
• stricter post-market surveillance to monitor faulty products,
• increased patient protection during clinical trials, and
• added data requirements for study sponsors.
Preparing for the new rules
Device manufacturers should allocate time and resources to adapt to stricter, more complex requirements.
The new MDR measures include incorporating unique device identifiers (UDI) on all medical device labels. The UDI and other pertinent medical data will be accessed through a revamped European Database for Medical Devices (Eudamed).
In spite of the official timeframes for mandatory implementation, changes already have begun to affect Europe’s Notified Body sector. Manufacturers are likely to have fewer Notified Bodies to choose from and to partner with for CE Mark certification, quality system inspections, and related functions going forward.
Effects On European Authorized Representatives
For foreign manufacturers that have no physical presence in Europe, establishing a partnership with a European Authorized Representative is crucial (and required) for CE Mark certification and successful commercialization.
Authorized Representatives act as liaisons between foreign manufacturers and national Competent Authorities. Authorized Representatives play a major role in dealing with both premarket and postmarket compliance and regulatory issues on behalf of manufacturing clients, and that role will continue under the new MDR and IVDR.
However, one change under the new regulations may have significant ramifications for how quickly or easily foreign manufacturers may find and establish partnerships with Authorized Representatives. Namely, the MDR will hold Authorized Representatives jointly liable, along with manufacturers, in cases where devices sold on the European market are found to be defective.
This change has major implications for foreign manufacturers, as firms offering Authorized Representative services can certainly expect more thorough due diligence and scrutiny from potential Authorized Representative partners who now will incur liability they did not have before. Foreign manufacturers also may undergo additional monitoring by their Authorized Representatives for postmarket compliance.